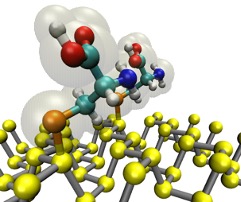
Since the synthesis of graphene in 2004, an extensive study of carbon-based and non-carbon-based 2-D materials has been carried inside the scientific community. 2D materials have shown outstanding chemical and physical properties due to quantum confinement, which has led to possible applications in catalysis, sensors, coatings, thermoelectric materials, and more. Among the non-carbon-based 2D materials, phosphorene has generated great attention due to its puckered structure with free pair of electrons, high electronic and thermal mobility, and its tunable direct bandgap.
In order to exploit phosphorene as an electronic, thermal, and thermoelectric material, chemical functionalization is desired to be computationally studied. Exploring the covalent functionalization with molecules of different functional groups can provide the opportunity for extended modulations and tuning of physical properties, giving access to the combination of the intrinsic properties of phosphorene with those of the functional groups, and serve as a basis of new material applications and hybrid heterostructures. [1]
The Chair of Materials Science and Nanotechnology is looking for a highly motivated student to complete her/his master thesis project in the project “Tuning the Electronic and Thermal Properties of Phosphorene by Chemical Functionalization”.
The student will:

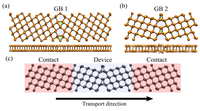
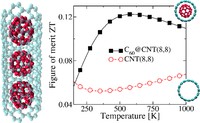
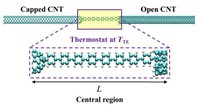
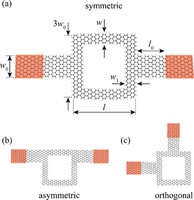
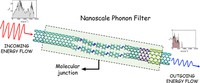
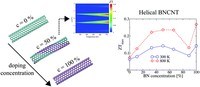
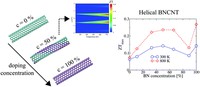
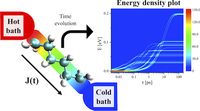

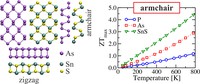
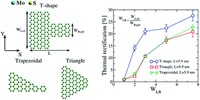
Since the synthesis of graphene in 2004, an extensive study of carbon-based and non-carbon-based 2-D materials has been carried inside the scientific community. 2D materials have shown outstanding chemical and physical properties due to quantum confinement, which has led to possible applications in catalysis, sensors, coatings, thermoelectric materials, and more. Among the non-carbon-based 2D materials, phosphorene has generated great attention due to its puckered structure with free pair of electrons, high electronic and thermal mobility, and its tunable direct bandgap.
In order to exploit phosphorene as an electronic, thermal, and thermoelectric material, chemical functionalization is desired to be computationally studied. Exploring the covalent functionalization with molecules of different functional groups can provide the opportunity for extended modulations and tuning of physical properties, giving access to the combination of the intrinsic properties of phosphorene with those of the functional groups, and serve as a basis of new material applications and hybrid heterostructures. [1]
The Chair of Materials Science and Nanotechnology is looking for a highly motivated student to complete her/his master thesis project in the project “Tuning the Electronic and Thermal Properties of Phosphorene by Chemical Functionalization”.
The student will:


