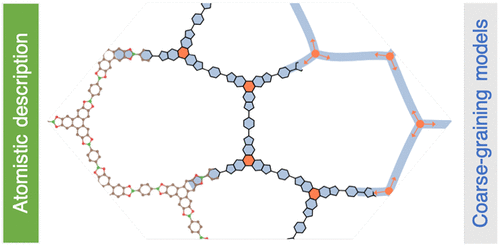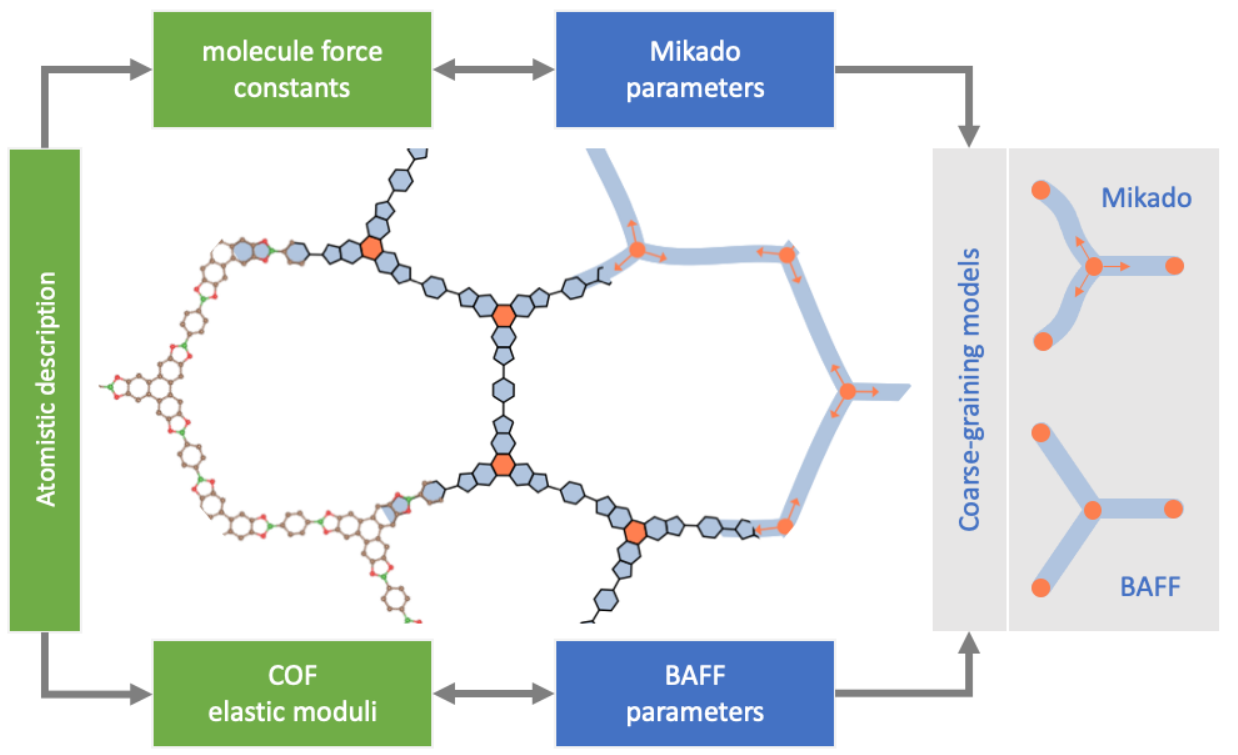
Covalent-Organic Frameworks (COFs) are crystalline porous materials that are based on organic monomeric units, so called building blocks. As a multitude of different building blocks can be combined in reticular chemistry, manifold different porous structures with tailored properties have been synthesized in recent years. Through recent experimental progress, monolayer COF materials have been synthesized, providing a new class of 2D materials. From a computational point of view, however, accurately calculating properties of these materials is computationally demanding as the unit cells of COFs are usually very big. In this work we calculate mechanical properties for a set of 2D COFs and compare results of density functional based tight binding (DFTB) with classical force-fields. We show how force-fields can be very useful for mechanical property calculation, how their accuracy can be improved, and typical fallacies for 2D porous materials. Furthermore, we introduce models to predict mechanical properties from the properties of the monomeric building blocks. This paves the way for multiscale modeling, high-throughput calculations, and materials prediction with properties on demand.
Covalent-Organic Frameworks (COFs) are crystalline porous materials that are based on organic monomeric units, so called building blocks. As a multitude of different building blocks can be combined in reticular chemistry, manifold different porous structures with tailored properties have been synthesized in recent years. Through recent experimental progress, monolayer COF materials have been synthesized, providing a new class of 2D materials. From a computational point of view, however, accurately calculating properties of these materials is computationally demanding as the unit cells of COFs are usually very big. In this work we calculate mechanical properties for a set of 2D COFs and compare results of density functional based tight binding (DFTB) with classical force-fields. We show how force-fields can be very useful for mechanical property calculation, how their accuracy can be improved, and typical fallacies for 2D porous materials. Furthermore, we introduce models to predict mechanical properties from the properties of the monomeric building blocks. This paves the way for multiscale modeling, high-throughput calculations, and materials prediction with properties on demand.