A local-orbital based ab initioapproach to obtain the Green function for large heterogeneous systems is developed. First a Green function formalism is introduced based on exact diagonalization. Then the self energy is constructed from an incremental scheme, rendering the procedure feasible, while at the same time physical insight into different local correlation contributions is obtained. Subsequently the Green function is used in the frame of the Landauer theory and the wide band approximation to calculate the electronic transmission coefficient across molecular junctions. The theory is applied to meta- and para-ditholbenzene linked to gold electrodes and various correlation contributions are analyzed.
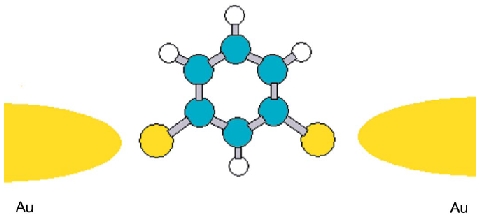
A local-orbital based ab initioapproach to obtain the Green function for large heterogeneous systems is developed. First a Green function formalism is introduced based on exact diagonalization. Then the self energy is constructed from an incremental scheme, rendering the procedure feasible, while at the same time physical insight into different local correlation contributions is obtained. Subsequently the Green function is used in the frame of the Landauer theory and the wide band approximation to calculate the electronic transmission coefficient across molecular junctions. The theory is applied to meta- and para-ditholbenzene linked to gold electrodes and various correlation contributions are analyzed.
