Despite great efforts in the experimental and theoretical investigation of charge migration in DNA oligomers, no unified explanation of the microscopic mechanisms governing charge transfer/transport has been achieved up to date. The complexity of this bio-molecule as well as the variety of factors that influence charge transfer/transport (solvent, base dynamics, nucleobase sequence, metal-molecule contact) make the available experimental data difficult to analyze unambiguously. Thus, theory and computation play a prominent role by guiding experimental studies and providing them with well-defined predictions concerning the role of all these factors. A fundamental problem in such systems is the strong coupling of electronic and structural degrees of freedom, which makes a treatment based on standard perturbation theory unfeasible in general. This review focuses on various methodological approaches to which the authors have strongly contributed. The advantage of these approaches relies on an efficient combination of accurate electronic structure calculations, classical molecular dynamics, and charge transport approaches, to describe charge migration in complex (bio)molecular systems.
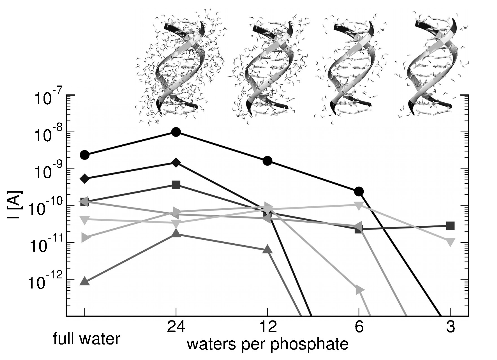
Despite great efforts in the experimental and theoretical investigation of charge migration in DNA oligomers, no unified explanation of the microscopic mechanisms governing charge transfer/transport has been achieved up to date. The complexity of this bio-molecule as well as the variety of factors that influence charge transfer/transport (solvent, base dynamics, nucleobase sequence, metal-molecule contact) make the available experimental data difficult to analyze unambiguously. Thus, theory and computation play a prominent role by guiding experimental studies and providing them with well-defined predictions concerning the role of all these factors. A fundamental problem in such systems is the strong coupling of electronic and structural degrees of freedom, which makes a treatment based on standard perturbation theory unfeasible in general. This review focuses on various methodological approaches to which the authors have strongly contributed. The advantage of these approaches relies on an efficient combination of accurate electronic structure calculations, classical molecular dynamics, and charge transport approaches, to describe charge migration in complex (bio)molecular systems.

