The power conversion efficiency of state of the art organic photovoltaics is predicted to be limited at about 15%. This limit can be increased by an improved charge carrier mobility and by a lower exciton binding energy. In order to achieve this, we suggest a concept based on organic triads, comprising a donor, spacer, and acceptor submolecule. On the basis of time dependent density functional theory (TD-DFT) simulations we investigate the lowest excited state of the well-known Carotenoid-Porphyrin-C60 triad and obtain a calculated exciton binding energy of 25 meV, justifying the experimentally observed and reported separation of photo generated charges. Further, we introduce a new triad with the ability to not only improve and control the separation process, but also to improve the charge carrier transport properties. We used molecular dynamics (MD) simulations to optimize the geometry of a cluster of 25 triads. From this organic cluster, we picked one triad with its four neighbors, again calculated the exciton binding energy of this structure and obtained 39 meV. We conclude that the exciton binding energy is stable for a variety of different basis set and functionals, and does not significantly change if one or a cluster of five triads is simulated. This stable behavior occurs, since changes in the wave functions do not significantly influence the exciton binding energy, as long the distance between the positive and negative charge remains the same. For photovoltaic applications and based on organic materials with a dielectric constant of about four, we suggest the use of spacer molecules larger than two nanometers.
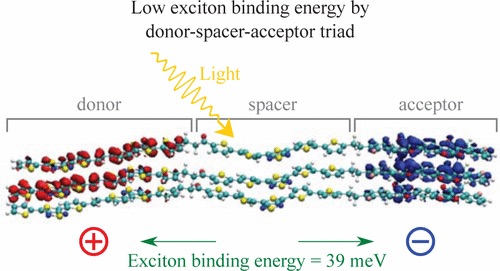
The power conversion efficiency of state of the art organic photovoltaics is predicted to be limited at about 15%. This limit can be increased by an improved charge carrier mobility and by a lower exciton binding energy. In order to achieve this, we suggest a concept based on organic triads, comprising a donor, spacer, and acceptor submolecule. On the basis of time dependent density functional theory (TD-DFT) simulations we investigate the lowest excited state of the well-known Carotenoid-Porphyrin-C60 triad and obtain a calculated exciton binding energy of 25 meV, justifying the experimentally observed and reported separation of photo generated charges. Further, we introduce a new triad with the ability to not only improve and control the separation process, but also to improve the charge carrier transport properties. We used molecular dynamics (MD) simulations to optimize the geometry of a cluster of 25 triads. From this organic cluster, we picked one triad with its four neighbors, again calculated the exciton binding energy of this structure and obtained 39 meV. We conclude that the exciton binding energy is stable for a variety of different basis set and functionals, and does not significantly change if one or a cluster of five triads is simulated. This stable behavior occurs, since changes in the wave functions do not significantly influence the exciton binding energy, as long the distance between the positive and negative charge remains the same. For photovoltaic applications and based on organic materials with a dielectric constant of about four, we suggest the use of spacer molecules larger than two nanometers.

