A surface-adsorbed molecule is contacted with the tip of a scanning tunneling microscope (STM) at a pre-defined atom. On tip retraction, the molecule is peeled off the surface. During this experiment, a two-dimensional differential conductance map is measured on the plane spanned by the bias voltage and the tip-surface distance. The conductance map demonstrates that tip retraction leads to mechanical gating of the molecular wire in the STM junction. The experiments are compared with a detailed ab initio simulation. We find that density functional theory (DFT) in the local density approximation (LDA) describes the tip-molecule contact formation and the geometry of the molecular junction throughout the peeling process with predictive power. However, a DFT-LDA-based transport simulation following the non-equilibrium Green functions (NEGF) formalism fails to describe the behavior of the differential conductance as found in experiment. Further analysis reveals that this failure is due to the mean-field description of electron correlation in the local density approximation. The results presented here are expected to be of general validity and show that, for a wide range of common wire configurations, simulations which go beyond the mean-field level are required to accurately describe current conduction through molecules. Finally, the results of the present study illustrate that well controlled experiments and concurrent ab initio transport simulations that systematically sample a large configuration space of molecule-electrode couplings allow the unambiguous identification of correlation signatures in experiment.
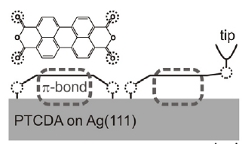
A surface-adsorbed molecule is contacted with the tip of a scanning tunneling microscope (STM) at a pre-defined atom. On tip retraction, the molecule is peeled off the surface. During this experiment, a two-dimensional differential conductance map is measured on the plane spanned by the bias voltage and the tip-surface distance. The conductance map demonstrates that tip retraction leads to mechanical gating of the molecular wire in the STM junction. The experiments are compared with a detailed ab initio simulation. We find that density functional theory (DFT) in the local density approximation (LDA) describes the tip-molecule contact formation and the geometry of the molecular junction throughout the peeling process with predictive power. However, a DFT-LDA-based transport simulation following the non-equilibrium Green functions (NEGF) formalism fails to describe the behavior of the differential conductance as found in experiment. Further analysis reveals that this failure is due to the mean-field description of electron correlation in the local density approximation. The results presented here are expected to be of general validity and show that, for a wide range of common wire configurations, simulations which go beyond the mean-field level are required to accurately describe current conduction through molecules. Finally, the results of the present study illustrate that well controlled experiments and concurrent ab initio transport simulations that systematically sample a large configuration space of molecule-electrode couplings allow the unambiguous identification of correlation signatures in experiment.


