The dynamics of a PTCDA-Ag(111) molecular junction has been investigated experimentally and theoretically. Switching between two conductance states was observed by low-temperature scanning tunneling microscopy, and was dependent on the tip-substrate distance and the applied bias, but was less sensitive to the bias polarity. Using a minimal model Hamiltonian approach combined with density-functional calculations, the switching was shown to be related to the scattering of electrons tunneling through the junction, which progressively excite the relevant chemical bond. Depending on the direction in which the molecule switches, different molecular orbitals are shown to dominate the transport and thus the vibrational heating process. This in turn can dramatically affect the behavior of the switching rate, demonstrating the importance of modelling the DOS realistically.
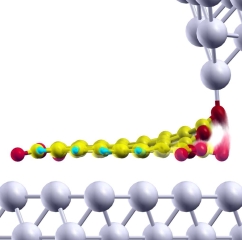
The dynamics of a PTCDA-Ag(111) molecular junction has been investigated experimentally and theoretically. Switching between two conductance states was observed by low-temperature scanning tunneling microscopy, and was dependent on the tip-substrate distance and the applied bias, but was less sensitive to the bias polarity. Using a minimal model Hamiltonian approach combined with density-functional calculations, the switching was shown to be related to the scattering of electrons tunneling through the junction, which progressively excite the relevant chemical bond. Depending on the direction in which the molecule switches, different molecular orbitals are shown to dominate the transport and thus the vibrational heating process. This in turn can dramatically affect the behavior of the switching rate, demonstrating the importance of modelling the DOS realistically.


