Semi-empirical quantum-mechanical (QM) methods have become valuable tools for studying complex (bio)molecular systems due to their balance between computational efficiency and accuracy. A key aspect of these methods is their parameterization, which not only governs the reliability of the results but also provides an opportunity to enhance their overall performance. In our previous work [J. Phys. Chem. Lett., 2021, 11, 16], we advanced the third-order semi-empirical density functional tight-binding (DFTB3) method for computing multiple properties of small molecules by developing the machine learning (ML) potential NNrep to bridge the gap between DFTB3 electronic components and those of the hybrid DFT-PBE0 functional. To overcome the limitations of NNrep, we introduce the EquiDTB framework, which leverages physics-inspired equivariant neural networks (NN) to parameterize scalable and transferable many-body ΔTB potentials, replacing the standard pairwise DFTB repulsive potential. This advancement extends the applicability of our ML-corrected DFTB approach to larger molecules and non-covalent systems (including only C, N, O, and H atoms), going beyond the chemical space represented in the training QM datasets. The enhanced performance of EquiDTB over the standard TB methods is demonstrated by the accurate computation of the atomic forces of S66x8 molecular dimers, as well as their interaction energies. Moreover, EquiDTB can be effectively employed to explore the potential energy surfaces of large and flexible drug-like molecules—for example, to determine the minimum energy path between isomers, analyze structural transitions during dynamical simulations, compute vibrational modes, and investigate energetic rankings. The performance for single molecules slightly decreases when the DFTB electronic energy is reduced to first-order but remains superior to standard TB methods. Our work thus demonstrates that an optimal integration of an equivariant NN with QM datasets can advance the DFTB method while maintaining high efficiency, paving the way for reliable (bio)molecular simulations.
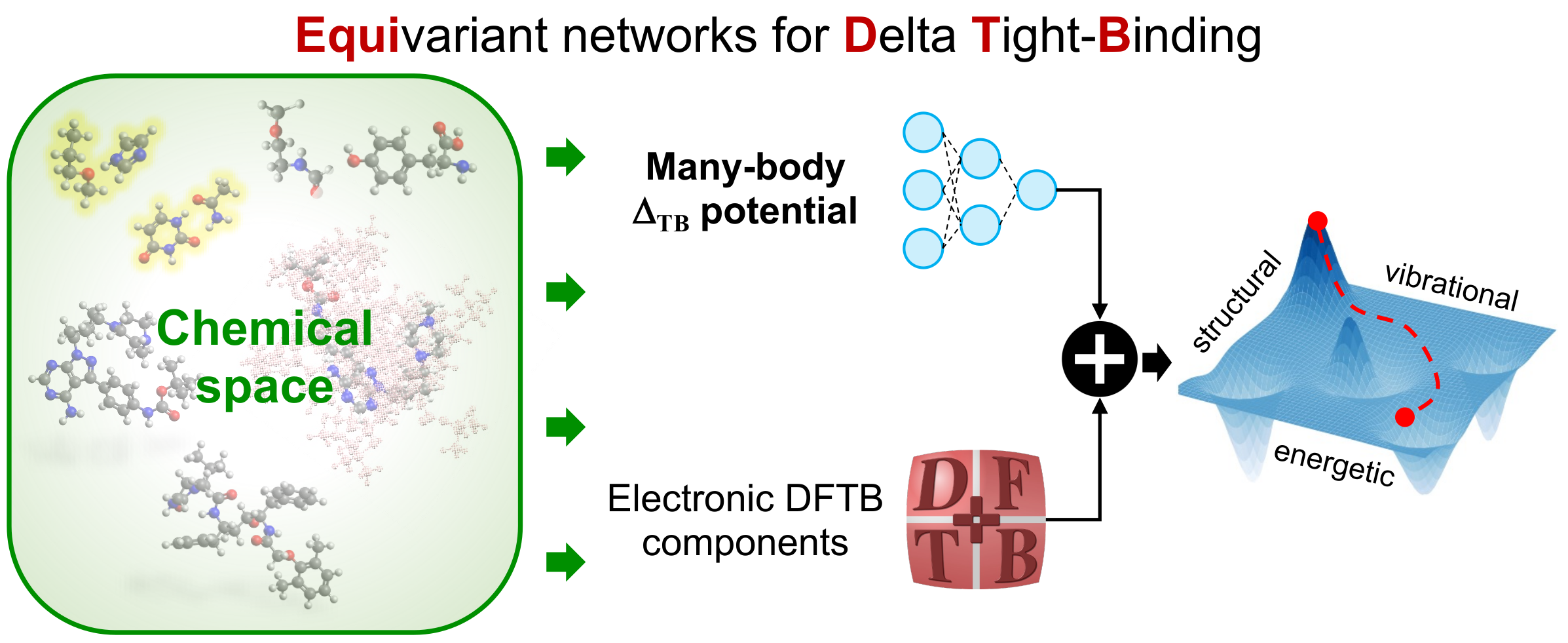
Semi-empirical quantum-mechanical (QM) methods have become valuable tools for studying complex (bio)molecular systems due to their balance between computational efficiency and accuracy. A key aspect of these methods is their parameterization, which not only governs the reliability of the results but also provides an opportunity to enhance their overall performance. In our previous work [J. Phys. Chem. Lett., 2021, 11, 16], we advanced the third-order semi-empirical density functional tight-binding (DFTB3) method for computing multiple properties of small molecules by developing the machine learning (ML) potential NNrep to bridge the gap between DFTB3 electronic components and those of the hybrid DFT-PBE0 functional. To overcome the limitations of NNrep, we introduce the EquiDTB framework, which leverages physics-inspired equivariant neural networks (NN) to parameterize scalable and transferable many-body ΔTB potentials, replacing the standard pairwise DFTB repulsive potential. This advancement extends the applicability of our ML-corrected DFTB approach to larger molecules and non-covalent systems (including only C, N, O, and H atoms), going beyond the chemical space represented in the training QM datasets. The enhanced performance of EquiDTB over the standard TB methods is demonstrated by the accurate computation of the atomic forces of S66x8 molecular dimers, as well as their interaction energies. Moreover, EquiDTB can be effectively employed to explore the potential energy surfaces of large and flexible drug-like molecules—for example, to determine the minimum energy path between isomers, analyze structural transitions during dynamical simulations, compute vibrational modes, and investigate energetic rankings. The performance for single molecules slightly decreases when the DFTB electronic energy is reduced to first-order but remains superior to standard TB methods. Our work thus demonstrates that an optimal integration of an equivariant NN with QM datasets can advance the DFTB method while maintaining high efficiency, paving the way for reliable (bio)molecular simulations.


