In this work, we propose computational strategies to improve the performance of the machine learning (ML)-augmented semi-empirical method EquiDTB, which is based on the density functional tight-binding (DFTB) formalism. To do this, we develop many-body ∆TB potentials using the state-of-the-art equivariant neural network architecture MACE. For training, we use structural and property data from ≈4.1 million equilibrium and non-equilibrium conformations of small drug-like molecules containing up to seven heavy atoms (C, N, and O). We explore two optimization strategies. The first one omits the second- and third-order electronic terms in the DFTB formalism, thereby eliminating the need for self-consistent charge calculations. Whereas the second one introduces an iterative training scheme that reduces the number of training conformations. Our results show that the optimized ∆TB potentials retain good accuracy and transferability, although their prediction errors in validation benchmarks are slightly higher than those of the original EquiDTB model. Importantly, the optimized models achieve better computational efficiency. Overall, this work provides a systematic assessment of optimization strategies that can guide the development of more efficient and reliable data-driven electronic structure methods for molecular simulations.
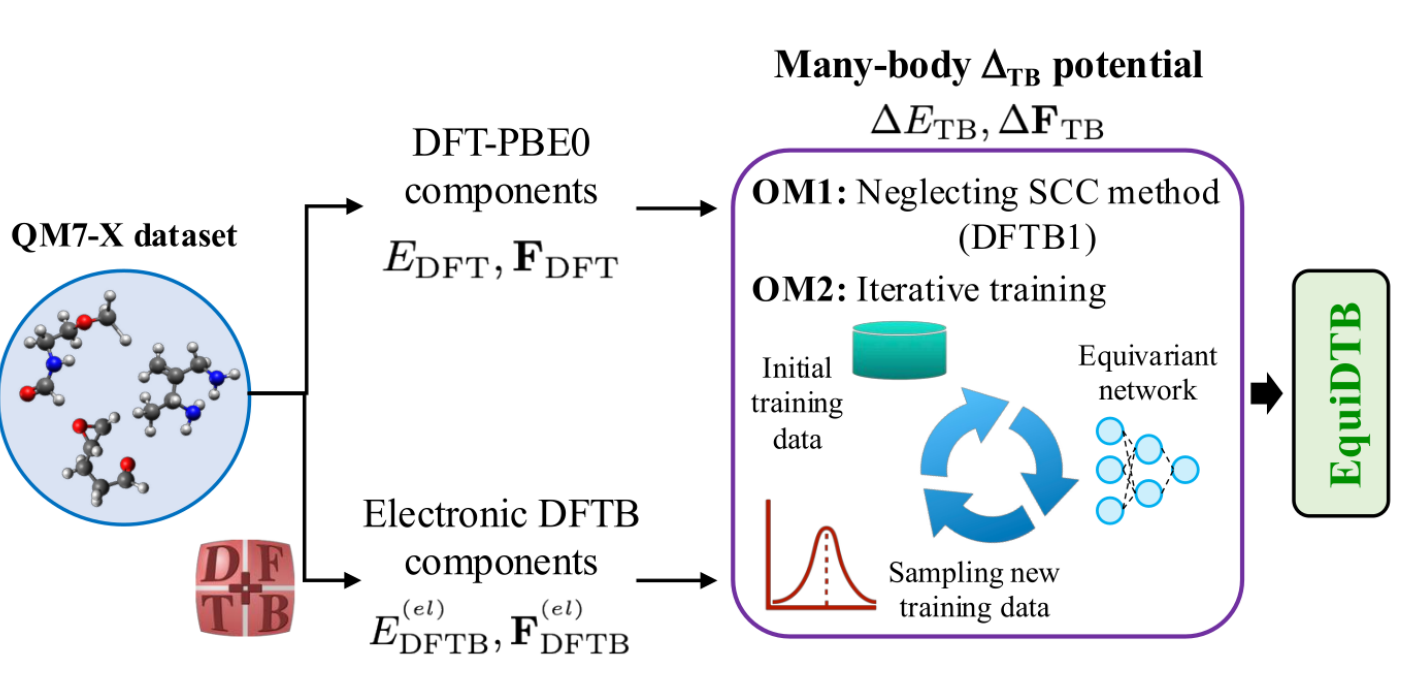
In this work, we propose computational strategies to improve the performance of the machine learning (ML)-augmented semi-empirical method EquiDTB, which is based on the density functional tight-binding (DFTB) formalism. To do this, we develop many-body ∆TB potentials using the state-of-the-art equivariant neural network architecture MACE. For training, we use structural and property data from ≈4.1 million equilibrium and non-equilibrium conformations of small drug-like molecules containing up to seven heavy atoms (C, N, and O). We explore two optimization strategies. The first one omits the second- and third-order electronic terms in the DFTB formalism, thereby eliminating the need for self-consistent charge calculations. Whereas the second one introduces an iterative training scheme that reduces the number of training conformations. Our results show that the optimized ∆TB potentials retain good accuracy and transferability, although their prediction errors in validation benchmarks are slightly higher than those of the original EquiDTB model. Importantly, the optimized models achieve better computational efficiency. Overall, this work provides a systematic assessment of optimization strategies that can guide the development of more efficient and reliable data-driven electronic structure methods for molecular simulations.


