
Methods for materials modelling on different length scales range from ab-initio DFT calculations of the electronic structure and of chemical reactions up to simulations of diffusion processes and the determination of macroscopic material properties of complex materials within the framework of continuum models by applying finite difference and finite element techniques.

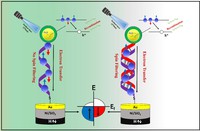
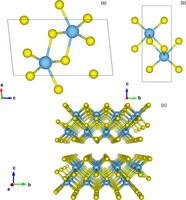
Simulation/Modelling of chirality induced spin selectivity (CISS)
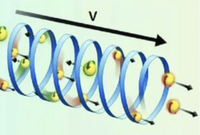
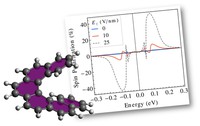
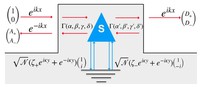
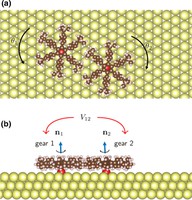
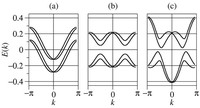
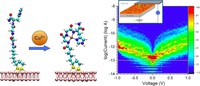
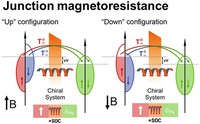
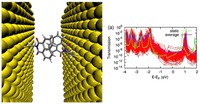
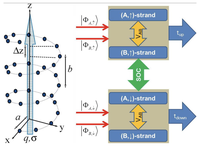
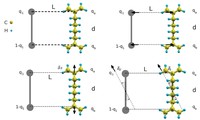
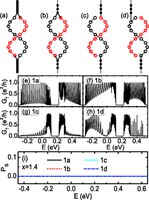
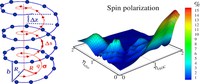
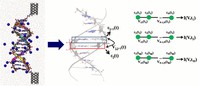
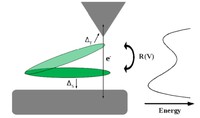
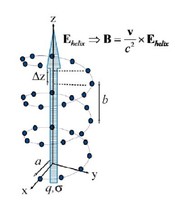
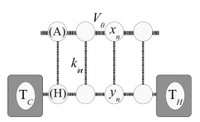
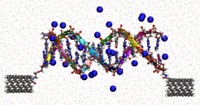
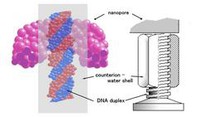
Recently, the chirality induced spin selectivity (CISS) effect has been discovered in which organic molecules serve as a spin filter. This is achieved due to the special property of chiral symmetry that couples the electron’s spin and its linear momentum so that the spin transmit one proffered spin. The CISS effect has been experimentally observed for several chiral/helical molecules such as DNA, oligopeptides, and chiral polymers. From theoretical point of view, the mechanism of spin selectivity through chiral molecules is not really understood. In this project we aim to elucidate the spin selectivity mechanisms through chiral molecules by simulation/modelling approaches.

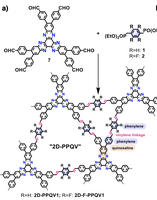
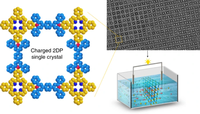
Charge Transport Characterization in 2D COFs
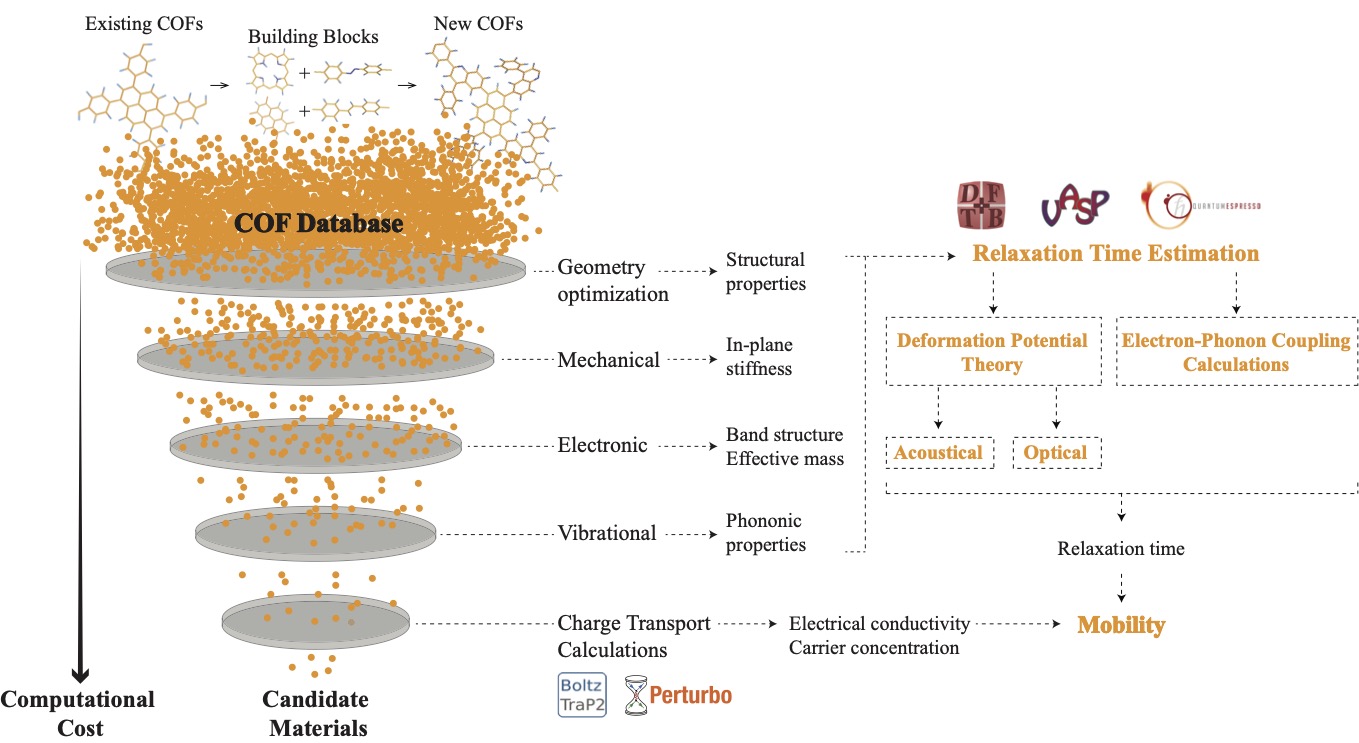
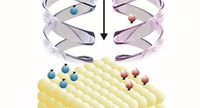
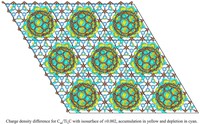
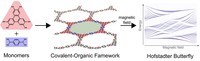
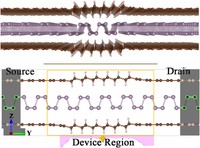
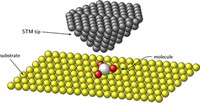
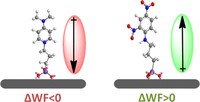
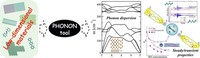
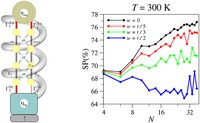
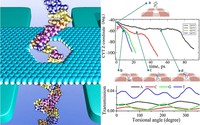
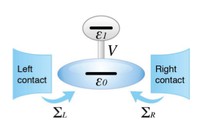
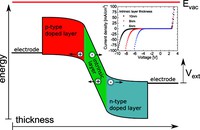
Two-dimensional (2D) covalent organic frameworks (COFs) have attracted great attention owing to their unique physical and chemical properties and as well as their potential for wide range of applications in electronics, sensing, energy storage and conversion. Diversity of molecular building blocks paves the way for the synthesis of novel 2D COFs with tunable and and predictable properties. Great variety of these materials necessitates a systematical way to analyze them. Despite the intensive studies on 2D COFs, charge transport properties of most of these materials are still unknown. In particular, their relation to molecular building block properties is a great challenge due to the combinatorial nature of reticular chemistry. We are investigating the structural and electronic properties of 2D COFs using density functional theory (DFT)-based first-principles approaches. We are modelling charge transport properties by combining the semiclassical Boltzmann transport theory and deformation potential theory (DPT) for acoustic and optical phonons. The results are bechmarked against state-of-the-art electron-phonon averaged (EPA) calculations.
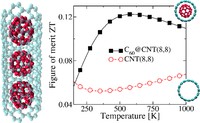
Tunning thermal and electronic properties of low dimensional materials
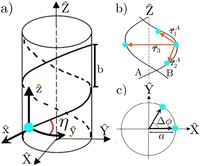
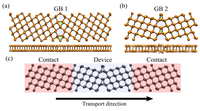
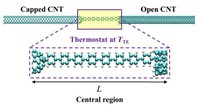
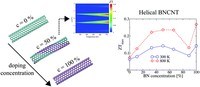
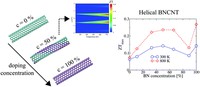
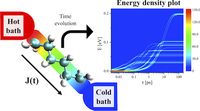
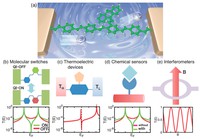
Since the synthesis of graphene in 2004, an extensive study of two dimensional materials have been carried inside the scientific community. 2D materials have shown outstanding chemical and physical properties due to quantum confinement, which has lead possible applications in catalysis, sensors, coatings, thermoelectric materials and more. One dimensional materials, like carbon nanotubes and silicon nanowires, do not stand behind since they also present particular properties like high electrical conductivity and thermal conductivity.
In order to exploit low dimensional materials as electronic, thermal and thermoelectric material, the effects of chemical functionalization, strain, structural defects and quantum confinement are computationally studied. In this wat one can provide the opportunity for extended modulations and tuning of physical properties and serve as a basis of new material applications and hybrid heterostructures.
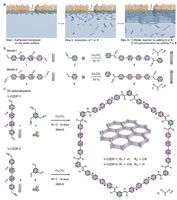
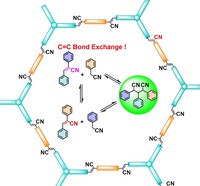
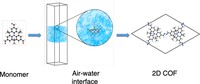
Investigation of bottom-up synthesis of 2D Polymers
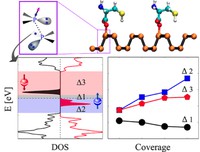
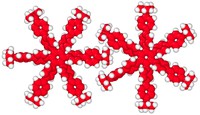
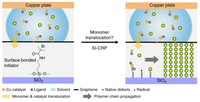
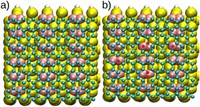
2D Polymers are versatile materials that have a great potential in areas like electronic applications, catalysis or membrane technology. However, controlling the thickness and crystallinity is still a great challenge today. Feng et al. recently developed a method which utilizes water-surfactant interface to enable a 2D templating effect. Through this, it has been possible to greatly decrease the thickness and increase the the size of the crystalline domains of such 2D Polymers. We investigate the molecular processes that guide the growth of monolayer and few-layer organic polymers and gain insight in how to better control the synthesis. For this, we use number of techniques like Monte Carlo, Molecular Dynamics or DFT(B).

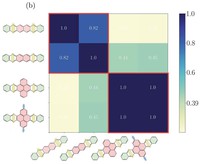
High-Throughput calculations of mechanical and electronic properties of 2D COFs
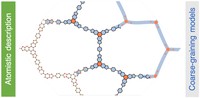
2D covalent-organic frameworks are made up of building blocks that are connected in a regular manner. By varying and combining different building block molecules, a vast amount of possible materials with different properties are accessible. In order to fabricate materials with tailored properties, however, it is necessary to either screen through many materials to find the right candidate or to understand the structure-property relationships in order to predict materials with specific properties. For this we use high-throughput workflows and machine-learning in combination with methods like classical molecular-mechanics and DFT(B).
Continuum Modeling
The general objective is to understand natural phenomena on a macroscopic scale. Our current activities focus on Li-Ion battery simulation at the continuum level. We use phenomenological physical models to simulate and predict the behavior of these cells under various conditions. More generally we are interested in all kinds of electrochemical processes, such as electrochemical deposition (i.e. Trenchfilling) and transport phenomena. To approach these scientific topics we use both commercial software tools and tailored in-house code in various programming or script languages.


















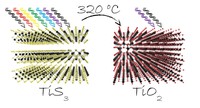
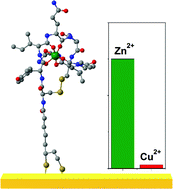
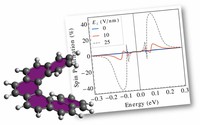
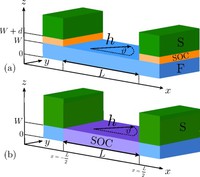
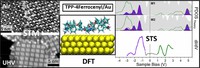
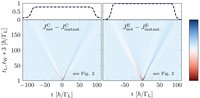
















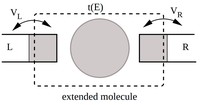


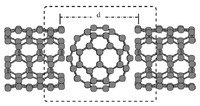
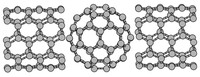
Methods for materials modelling on different length scales range from ab-initio DFT calculations of the electronic structure and of chemical reactions up to simulations of diffusion processes and the determination of macroscopic material properties of complex materials within the framework of continuum models by applying finite difference and finite element techniques.
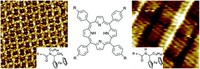
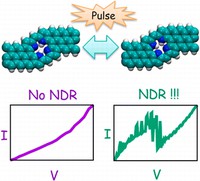
Simulation/Modelling of chirality induced spin selectivity (CISS)
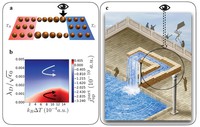
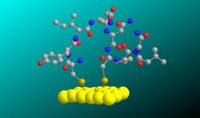
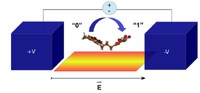
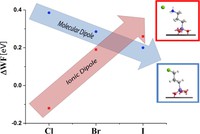
Recently, the chirality induced spin selectivity (CISS) effect has been discovered in which organic molecules serve as a spin filter. This is achieved due to the special property of chiral symmetry that couples the electron’s spin and its linear momentum so that the spin transmit one proffered spin. The CISS effect has been experimentally observed for several chiral/helical molecules such as DNA, oligopeptides, and chiral polymers. From theoretical point of view, the mechanism of spin selectivity through chiral molecules is not really understood. In this project we aim to elucidate the spin selectivity mechanisms through chiral molecules by simulation/modelling approaches.
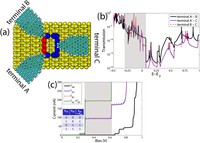
Charge Transport Characterization in 2D COFs
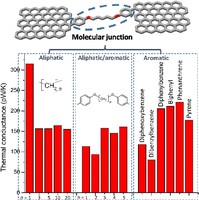
Two-dimensional (2D) covalent organic frameworks (COFs) have attracted great attention owing to their unique physical and chemical properties and as well as their potential for wide range of applications in electronics, sensing, energy storage and conversion. Diversity of molecular building blocks paves the way for the synthesis of novel 2D COFs with tunable and and predictable properties. Great variety of these materials necessitates a systematical way to analyze them. Despite the intensive studies on 2D COFs, charge transport properties of most of these materials are still unknown. In particular, their relation to molecular building block properties is a great challenge due to the combinatorial nature of reticular chemistry. We are investigating the structural and electronic properties of 2D COFs using density functional theory (DFT)-based first-principles approaches. We are modelling charge transport properties by combining the semiclassical Boltzmann transport theory and deformation potential theory (DPT) for acoustic and optical phonons. The results are bechmarked against state-of-the-art electron-phonon averaged (EPA) calculations.
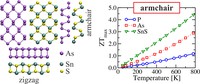
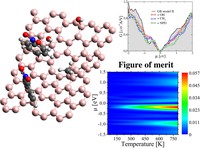
Tunning thermal and electronic properties of low dimensional materials
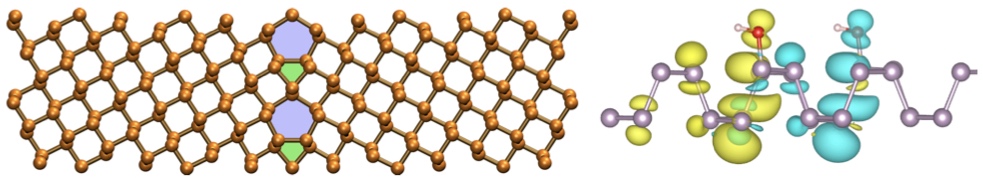
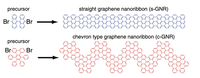
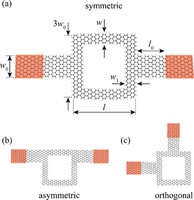
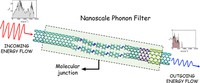
Since the synthesis of graphene in 2004, an extensive study of two dimensional materials have been carried inside the scientific community. 2D materials have shown outstanding chemical and physical properties due to quantum confinement, which has lead possible applications in catalysis, sensors, coatings, thermoelectric materials and more. One dimensional materials, like carbon nanotubes and silicon nanowires, do not stand behind since they also present particular properties like high electrical conductivity and thermal conductivity.
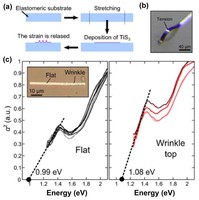
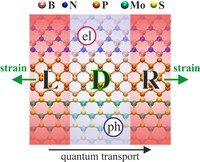
In order to exploit low dimensional materials as electronic, thermal and thermoelectric material, the effects of chemical functionalization, strain, structural defects and quantum confinement are computationally studied. In this wat one can provide the opportunity for extended modulations and tuning of physical properties and serve as a basis of new material applications and hybrid heterostructures.
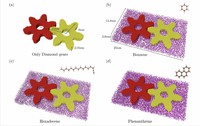
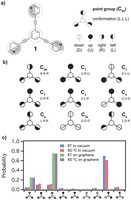
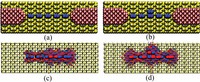
Investigation of bottom-up synthesis of 2D Polymers
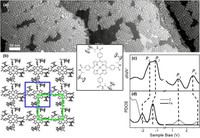
2D Polymers are versatile materials that have a great potential in areas like electronic applications, catalysis or membrane technology. However, controlling the thickness and crystallinity is still a great challenge today. Feng et al. recently developed a method which utilizes water-surfactant interface to enable a 2D templating effect. Through this, it has been possible to greatly decrease the thickness and increase the the size of the crystalline domains of such 2D Polymers. We investigate the molecular processes that guide the growth of monolayer and few-layer organic polymers and gain insight in how to better control the synthesis. For this, we use number of techniques like Monte Carlo, Molecular Dynamics or DFT(B).
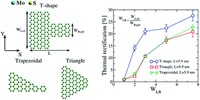
High-Throughput calculations of mechanical and electronic properties of 2D COFs
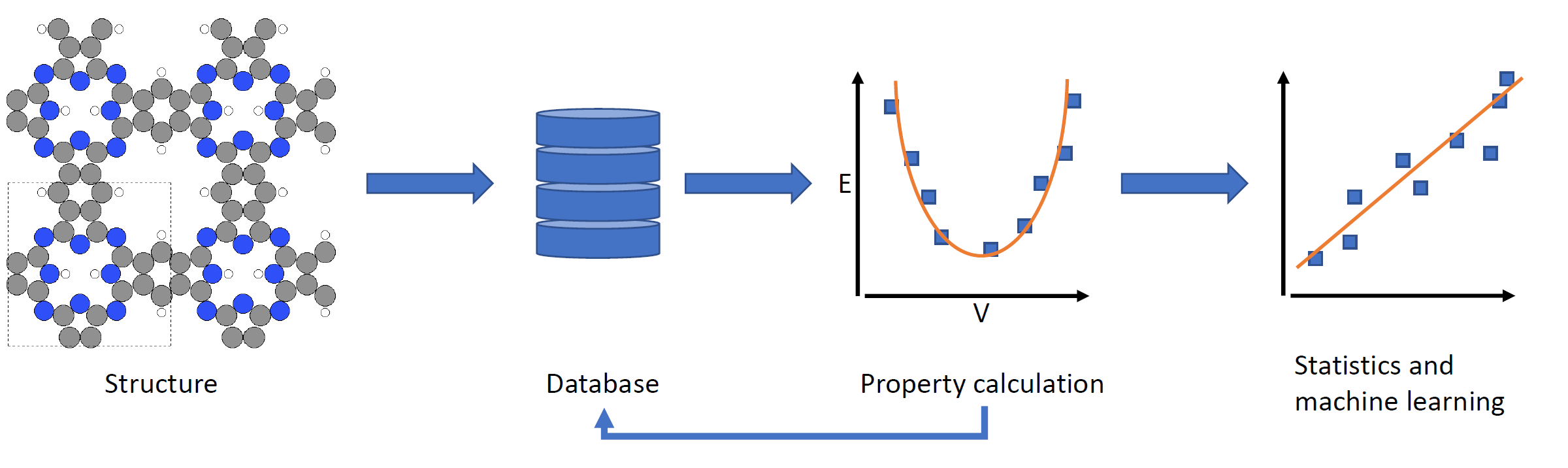
2D covalent-organic frameworks are made up of building blocks that are connected in a regular manner. By varying and combining different building block molecules, a vast amount of possible materials with different properties are accessible. In order to fabricate materials with tailored properties, however, it is necessary to either screen through many materials to find the right candidate or to understand the structure-property relationships in order to predict materials with specific properties. For this we use high-throughput workflows and machine-learning in combination with methods like classical molecular-mechanics and DFT(B).
Continuum Modeling
The general objective is to understand natural phenomena on a macroscopic scale. Our current activities focus on Li-Ion battery simulation at the continuum level. We use phenomenological physical models to simulate and predict the behavior of these cells under various conditions. More generally we are interested in all kinds of electrochemical processes, such as electrochemical deposition (i.e. Trenchfilling) and transport phenomena. To approach these scientific topics we use both commercial software tools and tailored in-house code in various programming or script languages.














